
文|财健道 尹莉娜
编辑|杨中旭
2月18日,港股上市企业荣昌生物(09995.HK)更新了其科创板IPO的招股书注册稿,其中包括其ADC药物维迪西妥单抗与美国FDA(美国药监局)沟通的进展。
对于这家由烟台传统中药企业荣昌制药“输血”十多年,与海归科学家房健民共同潜心培养起来的Biotech公司而言,维迪西妥单抗是其押注下一代肿瘤与免疫疗法ADC赛道的拳头产品,并以26亿美元的高价license-out美国ADC巨头Seagen,成为国产ADC海外授权第一例。
如果把PD-1单抗比作一颗精准发射的子弹,那么ADC药物,就好比在这颗子弹表面涂上剧毒,威力自然也将放大数倍。国外另一款ADC药物Lonca,在针对弥漫性大B细胞淋巴瘤的治疗中,成功让花费数十亿美金接受CAR-T细胞治疗失败后的患者,病情得到有效改善,重燃了对生命的希望。
但在10天前,中国创新药企业刚刚遭受了一场背后煎熬的“集体低潮”。
另一“港股18A公司”信达生物,其PD-1产品信迪利单抗作为首个国产PD-1赴美上市,遭到美国药监局肿瘤药物咨询委员会(FDA-ODAC)专家14:1的压倒性反对。
在国内竞争创新药内卷激烈、一上市就要“膝斩”“脚踝斩”进国谈的大环境下,仅凭国内市场收入,远无法支撑一家新药企业“车轮式”的烧钱投入现状。信达出海受挫,给所有在国内市场“吃不饱”,盼望着出海“赚美元”的生物医药企业,敲响了警钟。
事后,荣昌生物的首席医学官何如意对外表示:“如果药品有临床价值,能够满足未被满足的临床需求,即使临床试验数据都来自中国,去申报美国FDA仍然是可行的。”
一片愁云惨淡的本土新药市场,的确需要有人站出来振臂提气。而何如意在“下海”之前,曾在美国FDA工作逾17年,归国后还曾任中国药监局药品审评中心(CDE)的首席科学家,深谙中美两国在新药审批、临床试验要求上的差与池。
但他没有表明的是,荣昌生物的拳头产品ADC药物维迪西妥单抗,是否会像信迪利单抗一样,需要补充做“头对头试验”,方能拿到在美上市的许可券?
这个问题的背后,是中美业界所共同关注的——不仅“信达们”想知道,同样走在“仿创追赶”的道路上,继PD-1之后,下一代肿瘤治疗技术(如ADC、CAR-T)出海前景如何?国外数家“礼来们”也在琢磨,这些年费钱费力从中国引进的创新药项目,还值得在美国市场继续投入吗?
01 荣昌生物维迪西妥单抗,26亿美金开启ADC出海第一例
2014年,全球首款PD-1获批上市;2019年,首款国产PD-1面世,国内企业仅用5年时间实现了技术“跟随”。
而荣昌生物的维迪西妥单抗,也是我国首款国产ADC药物的获批在2021年,却比海外整整晚了21年时间(2000年全球首款ADC药物上市)。
这是因为,ADC药物是继化疗药物、小分子靶向药、单抗药物后的第四代肿瘤治疗技术。而其复杂性,也远远高于PD-1等单抗药物。

ADC药物,就是在业界熟知的单体药物基础上,用某种特定的连接物(Linker),以特定方式将小分子细胞毒药物(Payload)结合起来,从而达到1+1远大于2的效果。
如此一来,既能保持单抗药物的高特异性,又不丢失小分子药物的高活性,从而提高肿瘤药物的靶向性、减少毒副作用。
如果将单抗药物比作一颗精准发射的子弹,那么ADC药物所做的创新,就是为这颗子弹涂上剧毒,导致不仅其击中之处的肿瘤细胞死亡,还能通过“毒药”的扩散杀死周围的其他肿瘤细胞(医学上称为“旁观者效应”)。
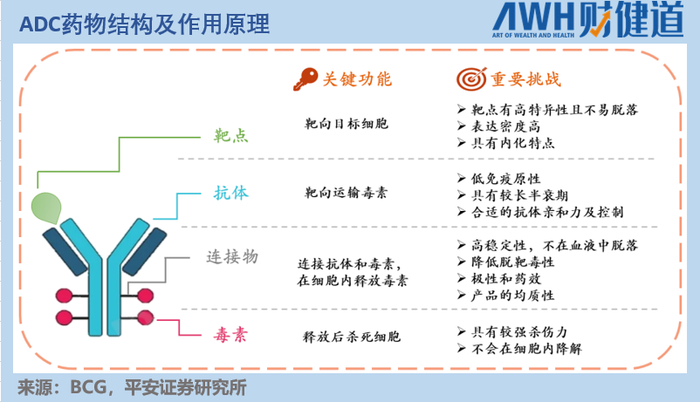
那么,问题来了——要选择什么样的单抗,作用于哪一靶点,携带怎样的Payload,用何种Linker,用什么方式联结起来,才能发挥最稳定且最好的效果呢?
失之毫厘,差之千里,这就是ADC这种明星药物的研发“磨人”之处。
目前布局的十几家本土药企中,产品走在最前面的,是荣昌生物的维迪西妥单抗。其在本土ADC中最早上市,并且还有1项适应症已进入上市审批(BLA),2项适应症正在进行注册性临床试验,另有3款储备产品已进入临床阶段。

2021年6月,其第一款ADC药物,也是公司的明星产品维迪西妥单抗获NMPA(国家药监局)批准上市,又在当年12月顺利进入医保,成功巩固先发优势,像极了当年医保谈判“先发降价”、次年产品销售额远超第二名君实生物近13个亿的信达生物。
但即便如此,如同当年“第一批吃到螃蟹”的信达一样,仅凭国内市场显然无法满足荣昌收回源源不断投入的研发成本。据荣昌生物预计,维迪西妥单抗2022年销售额为4-5亿元。而随着对新适应症的开发和产能的扩建等,其资金投入还将不断扩大。
海外市场,成为荣昌生物的必争之地。
好消息是,维迪西妥单抗获得了FDA突破性疗法认证,还以总计26亿美元的高价license-out美国ADC药物Seagen,成为国产ADC海外授权第一例。
但在研发进展上,根据荣昌生物不久前(2月18日)更新的科创板招股书注册稿,Seagen在2月14日与美国FDA开展了讨论,“美国FDA同意了维迪西妥单抗用于治疗尿路上皮癌的II期注册性临床试验方案,目前该项试验处于全球临床中心启动准备阶段,公司预计将于2022年4月份完成首例患者入组。”
若根据此次信达生物ODAC会议释放的信号来看,维迪西妥单抗很可能需要开展“头对头试验”,这是指在试验中将临床上已经使用的治疗药物作为对照组。
在肿瘤治疗中,相比以化疗为对照,与已上市的药物(如K药/O药)开展“头对头试验”,意味着花费的时间将大大延长,资金投入也非同一量级。此前业内猜测,补充头对头实验将耗费信达生物至少数千万美金,4-5年时间。
02 “头对头试验”成为赴美上市必需?
此前,国产创新药成功出海的经验并非为零。
在中国创新药产业飞速发展的十几年里,百济神州的BTK抑制剂泽布替尼,是迄今为止唯一一个成功敲开美国FDA大门获批上市的国产抗癌药。
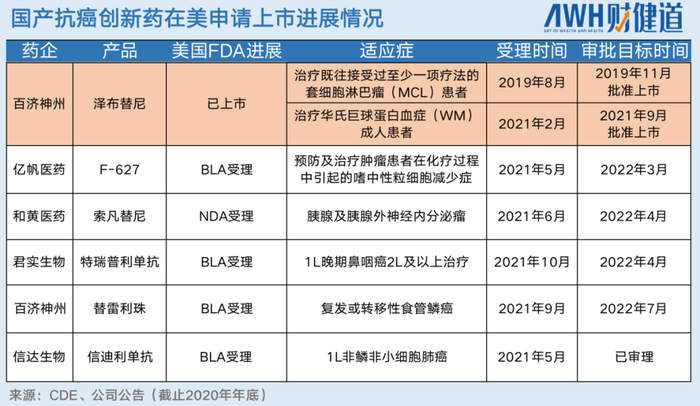
但该药物批准时也未开展针对伊布替尼的“头对头试验”(百济泽布替尼在美上市后,曾开展针对另一适应症华氏巨球蛋白血症的“头对头试验”,但与首个适应症套细胞淋巴瘤获批无关),由此产生一个疑问,“头对头试验”是否真的必需?
一位国内头部创新药企业准入人员告诉《财健道》,“有声音认为,FDA的评审规则变了,从泽布替尼到信迪利单抗,‘头对头试验’成为必须,但我对‘信达事件’的反思是,规则一直都很明确,‘头对头’不是关键,问题在于临床试验的设计和最终结果都足够有说服力,特别是要满足FDA的各项要求。”
目前,这一点仍无法盖棺定论。不过,回顾其上市前的临床试验,尽管二者获批前都未开展“头对头试验”,但泽布替尼与信达生物信迪利单抗的实验设计确实存在较多差异。
最基本的两点——
一是前者开展了包括澳洲在内的国际多中心临床试验,入组患者涵盖多个不同人种人群,而后者仅在国内开展实验,即单一人群患者。FDA认为,这意味着其无法排除信迪利单抗可能由于环境、人种的差异等,导致存在剂量或不良反应等的差别。
二是,后者的临床试验设计以无进展生存期(PFS)为临床终点,而FDA专家认为,应当以总生存期(OS)为临床终点,OS才是最可靠的“金标准”。
在中国市场的监管政策下,这样的试验设计并无不妥。根据CDE于2019年发布的《晚期非小细胞肺癌临床试验终点技术指导原则》,“随着治疗手段的丰富,OS不断延长增加了评价难度,因此单独PFS或PFS与OS共同终点可被接受作为初治晚期NSCLC(非小细胞肺癌)注册研究的主要终点。”
因而,目前所有已批的国产PD-1单抗的相关临床终点都是PFS,但海外则不同,近年来美国批准的NSCLC药物基本都以OS终点为主。
国内某知名肿瘤专科三甲医院淋巴瘤教授告诉《财健道》,对于信达PD-1赴美上市遭到反对的结果,早有预期。此前在另一项小众适应症的临床试验中,信达曾因“结果有效率和完全缓解率数据过高,数倍于行业认知”而遭到业内质疑,“谁信呢?”
03 欲速则不达临床试验合理合规是关键
换句话说,信达生物折戟FDA的原因可能在于,其过于着急想要拿到第一个在美上市的国产PD-1的批件,仅用国内试验数据匆匆到美国过堂,而“忽略”了美国FDA对创新药审批的多项要求。
回到荣昌生物的维迪西妥单抗。
丰硕创投研究总监莫灿龙告诉《财健道》:“任何一款创新药出海的成功与否,关键仍在于能否满足未满足的临床需求。要做到这点,要么在同类产品中或某一细分适应症上,速度处于前沿;要么风险获益比要优于同类产品或现有标准疗法。简单说,要么快,要么好。”。
所谓“快”与“好”,二者都是相对概念,需要对比同适应症的竞品来看。
维迪西妥单抗在美国瞄准的首个适应症,是尿路上皮癌。
而在这一适应症上,美国FDA已于2019年12月批准由安斯泰来/Seattle Genetics联合开发的ADC药物Enfortumab Vedotin(简称“EV”)用于治疗局部晚期或转移性尿路上皮癌。
此项批准所依据的临床试验纳入了125例患者,之前都接受了PD-1或PD-L1抑制剂和铂类化疗。试验结果表明,所有患者的客观缓解率(ORR)为44%,完全缓释率(CR)为12%,中位应答持续时间(DOR)为7.6个月。
而根据2021年ASCO大会上公布的最新数据,荣昌生物目前开展的II期临床研究RC48-C009,入组64例患者为接受过目前所有有效化疗(包括吉西他滨、铂类和紫杉醇类等)失败的患者。接受RC48-ADC治疗的ORR为50.0%,中位DOR为8.3个月。
若直接对比,会发现ORR和DOR2项关键试验结果数据,荣昌生物ADC均优于安斯泰来,但事实上,二者的试验区别有三:
一是,与信达的Orient-11一样,荣昌生物的国内试验未入组不同人种患者;二是其入组患者数量较少,仅为EV试验的1/2;三是EV试验入组患者均为已接受过PD-(L)1治疗的患者,与荣昌生物的化疗后患者相比,难度更大,也更符合伦理。
所谓符合伦理,即在有更好的治疗方法时,应当为患者提供这一选择,而非用更次的化疗方案或安慰剂作为对照组,即使这些患者只是试验人群。
此次信达ODAC会议上,也有多个声音明确提出相关质疑。
有提问者对信达生物在美国的合作伙伴礼来(Eli Lilly)发出质疑:“礼来对剥夺患者延长总体生存期的治疗的化疗组感到满意吗?礼来进行了多少次试验,剥夺了患者获得已知生存优势的治疗方法?”
一位在场专家也表示:“感到非常不舒服,因为已知疗法可以改善一年以上的平均生存率,而患者却没有得到它。”
由此来看,荣昌生物ADC药物开展国际多中心临床试验必不可少,并且合理、合规的试验设计和执行十分关键。
04 靶点扎堆,同质化设计国产ADC再陷PD-1式“内卷困境”?
国产ADC药物能否成功出海,归根结底,考验的还是新药研发和ADC技术产业化的综合实力。
莫灿龙向《财健道》表示,“截止2021年底,全球共有14款ADC药物上市,我国只有1款国产的ADC药物上市。毫不客气地说,过去我国ADC药物的发展程度是远远跟不上国外的。”
据《财健道》不完全统计,目前国内有产品进入临床阶段的国产ADC药物企业共13家。
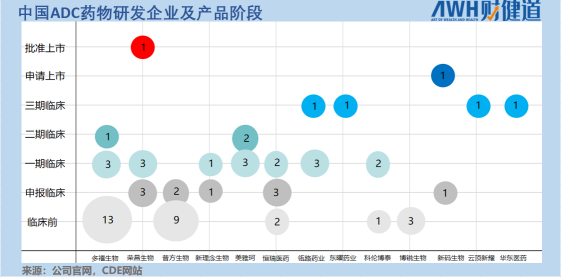
这些公司ADC药物的靶点设计基本集中在HER2和Trop2,其中进入临床试验阶段的HER2靶向ADC共有17个,几乎占据半壁江山;同时,布局Trop2靶点的ADC企业有11家。
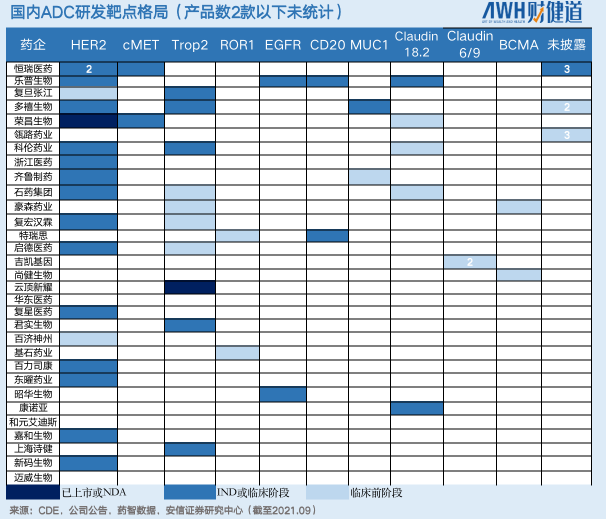
不得不说,从PD-1到ADC,国内新药研发靶点扎堆、结构设计同质化的现象依然在上演。
某头部机构医药分析师表示,但对比分析国内大部分ADC在研产品不难发现,其从单抗到linker、payload再到偶联方式的选择,都高度相似。即便已上市/进入临床后期的产品,也很难让人眼前一亮。“但ADC药物最大的魅力,正在于其结构存在着无数种排列组合的可能,因此更有可能诞生天马行空般的源头创新。”
05 第一三共EnhertuADC界“天花板”炼成背后的启示
在这方面,日本药企第一三共以一款颠覆性ADC药物提供了范例。
2019年,其开发的靶向HER2的ADC药物Enhertu(Trastuzumab Deruxtecan,研发代码DS-8201)获批上市。而后被Fierce Pharma评为最受期待的十大新药之首,并且被阿斯利康以69亿美元的总金额引进了在海外的商业化开发权益。
如同PD-1界的“K药”一般,Enhertu在临床疗效、稳定性、市场份额等各方面都成为ADC界的“天花板”。Evaluate Pharma曾预测,Enhertu在2024年的销售额将达到20亿美元。
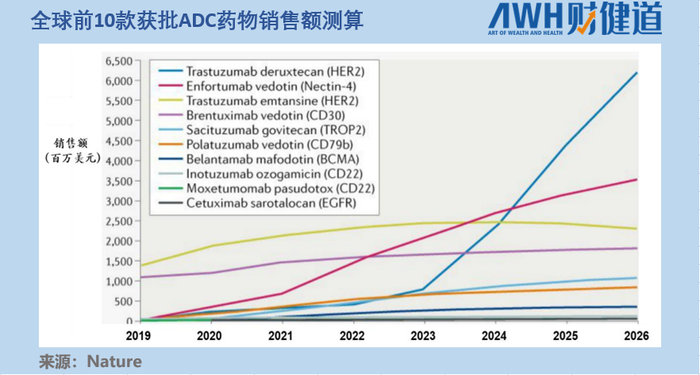
由于疗效突破显著和适应症丰富,据《Nature》文章测算,2020年以前上市的10款ADC药物,到2026年Enhertu(上图中蓝线代表Enhertu,其后期增速一骑绝尘)预计将以62亿美元的销售额位居第一位,占据约40%的市场份额。
这项药物最突出的过人之处在于其独有的linker技术,据报道,第一三共的研发团队当时合成了上百种linker,不断优化筛选后,Enhertu每个抗体上所能结合的细胞毒药物数量,史无前例地达到了8个。这为其带来的威力好比,当对手(其他ADC药物)的枪膛里只有3-5颗、甚至1颗子弹时,Enhertu有8颗子弹。
而第一三共开始做布局ADC项目的研发,始于2010年,除了百里挑一的linker,其对payload和抗体的筛选同样千锤百炼。到今天,经过20多年的持续耕耘,第一三共在ADC方面的主要成功药物也不过3款。其2021年发展计划显示,到2025年的未来5年里,第一三共预计将投入130亿美金在这3款ADC药物上,也就是说平均每款药物每年将花费8.6亿美金,这尚不包括在2021年之前10年的研发投入。
这恰恰提示了,刹那迸发而千古流芳的“创新”,与日复一日却不见天日的“积累”,就好比一枚硬币的正反面。研发成功的“好运气”,只留给持续深耕的企业。
也正因如此,莫灿龙认为,作为下一代肿瘤治疗技术,ADC药物的发展现在还不到见分晓的时刻。“随着政策、资本和人才的持续加码,ADC药物的国内外差距正急速缩小,处于快速追赶阶段。”

如果把“照搬式”的Me-too药物比作1.0阶段,那么在跟仿的同时,能够做出一些有临床价值的改善(Me-better),或者在已经发现还未得到充分验证的靶点进行尝试(Me-faster),便可以称为2.0阶段。再进一步发展,则是First-in-class级的“全球新”3.0阶段。
当前,国产ADC药物正处于从1.0到2.0的追赶时期。
莫灿龙表示,国产ADC药物的整体水平要想达到世界一流的水平,需要ADC药物的整条产业链,包括药物发现、临床前研究、生产制造、临床开发以及上市监管等均发展成熟。
最后,尽管Enhertu已经成为业界公认最为创新的ADC产品,但其仍有自己的缺陷,就是不良反应。
今年1月21日发表在《OncLive》的一项II期临床试验结果显示,126位使用Enhertu的患者中,有85.6%发生了3级或3级以上不良反应,而使用化疗的患者中的发生率为56.5%。而在更早前的一项其他临床研究中,还曾4例患者因间质性肺炎不良反应而致死的事件。
不良反应的问题表明,行业“天花板”,也仍有被突破的空间,创新研发,就是不断创造不可能。
参考文献
【1】对话原国家药监局首席科学家何如意:现在的任务是帮助中国创新药出海,深蓝观,2022.02.14
【2】 ADC赛道竞速赛:巨头药企引领,国内药企迎收获期,美柏医健,2021.11.22
【3】创新药研发专题系列——ADC:从模仿转型创新之路,平安证券,2021.07.22
【4】手握8弹头ADC“制导炸弹”,第一三共将如何布局中国肿瘤领域,E药经理人,2021.04.06
评论