
文 | 千芯科技董事长 陈巍
就在前几天,迄今为止参数最多、规模最大的蛋白质预测模型ESMFold被Meta官宣了,甚至有研究者宣称该模型又大又好,足以碾压Google在2021年推出的AlphaFold2。

▲ESMFold与通讯作者Meta AI的Alexander
这一消息着实让学术界和工业界震撼,要知道这些大的模型,无论训练还是使用,都得有妥妥的“钞能力”,如果模型越来越小,说不定就不需要更大算力的芯片了。(当然事实并非如此)甚至LeCun大牛都发推为ESMFold背书,称之为“Super-fast and accurate”。
从氨基酸序列预测蛋白质结构是自然科学中长期存在的重大挑战。在基于进化的算法中,AlphaFold2可以说是目前解决该问题最成功的。它通过在多序列输入、进化同源物对齐序列和可选结构模板上训练端到端神经网络,取得了突破性成就,大大加速了“生命元宇宙”的构建。
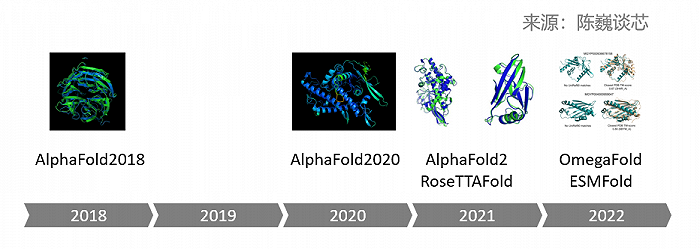
▲蛋白质预测AI大模型的进化
而Meta团队的ESMFold蛋白质模型只需要一个序列作为输入,该模型背后的团队由Meta AI(原Facebook AI)的资深研究科学家Alexander Rives主导。该团队专注于大规模蛋白质序列和结构数据的无监督表示学习模型研究。Alexander本人同时也是Fate Therapeutics、Syros Pharma、Kallyope的联合创始人,妥妥的科创家。
那ESMFold真的能碾压AlphaFold2吗?让我们先来回顾下什么是蛋白质结构预测,然后再深入分析ESMFold的网络结构。
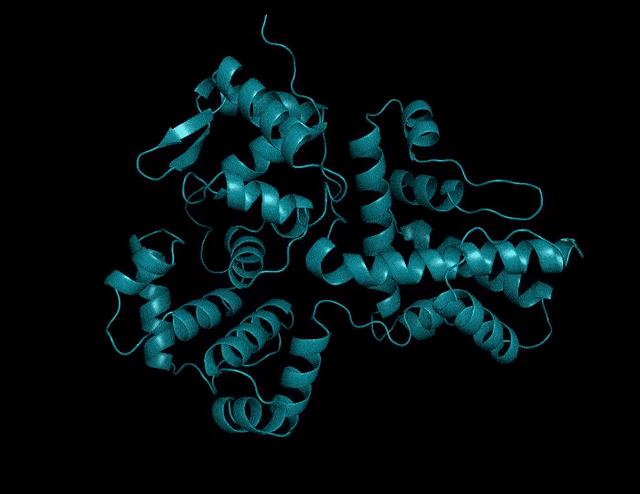
▲ESMFold预测的结构
论文链接:https://doi.org/10.1101/2022.07.20.500902
01.什么是蛋白质结构预测?
首先,蛋白质结构是指各种蛋白质分子的空间结构。由线性氨基酸组成的蛋白质,需要折叠(Fold)成特定的空间结构,才具有相应的生理活性和生物学功能。
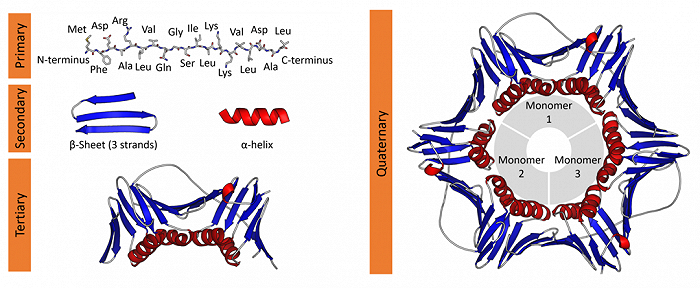
▲蛋白质的四级结构
蛋白质的分子结构可划分为四级,以描述其不同层级的特征:
蛋白质一级结构:组成蛋白质多肽链的线性氨基酸序列。
蛋白质二级结构:依靠不同氨基酸之间的C=O和N-H基团间的氢键形成的稳定结构,主要为α螺旋和β折叠。
蛋白质三级结构:通过多个二级结构元素在三维空间的排列所形成的一个蛋白质分子的三维结构。
蛋白质四级结构:用于描述由不同多肽链(亚基)间相互作用形成具有功能的蛋白质复合物分子。
我们所说的蛋白质结构预测(Protein Structure Prediction),就是指从蛋白质的氨基酸序列中预测蛋白质的三维结构。也就是说,从蛋白质的一级结构预测其折叠和二级、三级、四级结构。
DeepMind(Google旗下)的AlphaFold2在蛋白质结构预测大赛CASP 14中,对大部分蛋白质结构的预测与真实结构只差一个原子的宽度,达到接近冷冻电镜等复杂仪器检测的水平。这一巨大进步被Nature和Science选为2021年度十大科学突破。
根据不同的氨基酸和序列,蛋白质能折叠成的构型数量是一个天文数字,因此很难用常规方法进行蛋白质结构的准确预测。例如,目前实验的方法(例如冷冻电镜)至今才能解出10万的蛋白质结构。
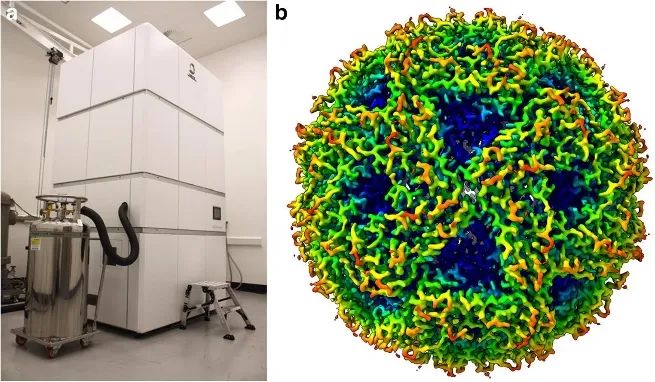
▲冷冻电镜及其图像
因此,使用AI的方法,加速对蛋白质结构的解析,分析其组成和功能,就成了生物界和医药界的争相推进的重要工作。
02.ESMFold的“魔幻效果”
ESMFold与AlphaFold2和RoseTTAFold对多序列输入的蛋白质结构预测具有相当的准确度。但ESMFold突出优势在于,其计算速度比AlphaFold2快一个数量级,能够在更有效的时间尺度上探索蛋白质的结构空间。
过去,AlphaFold2和RoseTTAFold在原子分辨率蛋白质结构预测问题上取得了突破性成功,但依赖于使用多序列比对(Multiple Sequence Alignment,简写为MSA)和相似蛋白质结构的模板来实现最优表现。
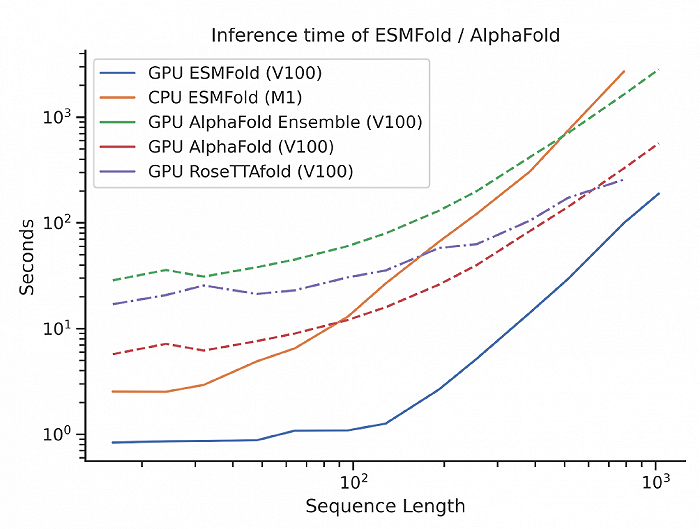
▲ESMFold模型具有比AlphaFold2更高的速度
ESMFold使用ESM-2学习的信息和表示来执行端到端的3D结构预测,特别是仅使用单个序列作为输入(AlphaFold2需要多序列输入),方便研究者在使用时通过模型缩放,将模型大小控制在数百万到数十亿量级参数。需要注意的是,随着模型大小的增加,可观察到预测准确性的持续提升。(还是“越大越准”)
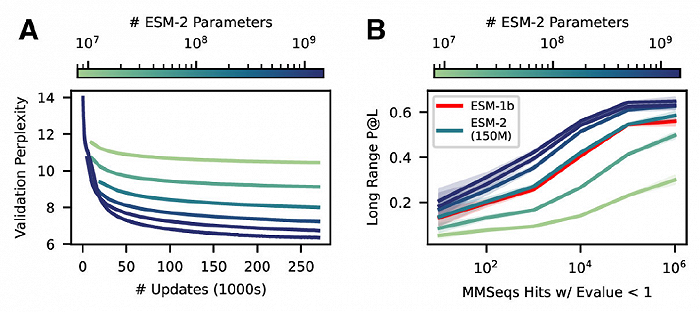
▲ESM-2模型随着参数量升高精度升高
由于ESMFold的预测速度比现有的其他原子分辨率蛋白质结构预测模型快一个数量级,因此ESMFold可以帮助快速构建蛋白质结构数据库。使用ESMFold,可以快速计算100万个预测结构,这些结构代表了蛋白质预测空间的不同子集,其中大多数没有注释的结构或功能。
而且ESMFold的大部分高置信度预测与已知的实验结构的相似度都很低,这表明了通过AI计算获得的基因组蛋白的结构新颖性。
值得注意的是,许多高置信度结构与UniRef90中的结构也具有低序列相似性,说明该模型具有超出其训练数据集的泛化能力,实现了基于结构的蛋白质功能预见能力。
据此,研究人员认为,ESMFold可以帮助理解那些超出现有认知的蛋白质结构。
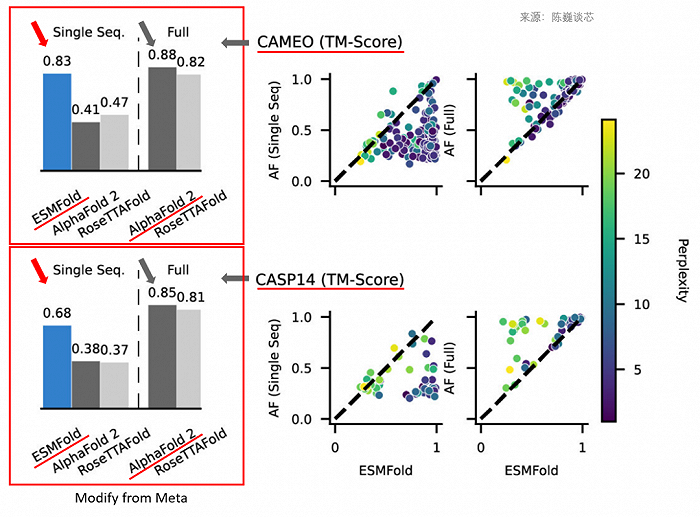
▲ESMFold在单序列输入时预测精度明显好于AlphaFold2
虽然ESMFold速度很高,精度也不错,特别是在单序列输入的时候精度明显好于AlphaFold2。但我们也要看到,ESMFold在多序列输入的情况下,其精度比AlphaFold2还是略有差距。
03.ESMFold网络结构
与AlphaFold2模型类似,ESMFold模型的架构也可以分为四部分:数据解析部分、编码器部分(Folding Trunk)、解码器部分(Structure Module)、循环部分(Recycling)。
ESMFold和AlphaFold2之间的一个关键区别是使用语言模型表示来消除对显式同源序列(以MSA的形式)作为输入的要求。
语言模型表示作为输入提供给ESMFold的折叠主干。通过将处理MSA的计算量大的Folding Block模块替换为处理序列的Tranformer模块来简化AlphaFold2中的Evoformer。这种简化或优化意味着ESMFold会比基于MSA的模型快得多。
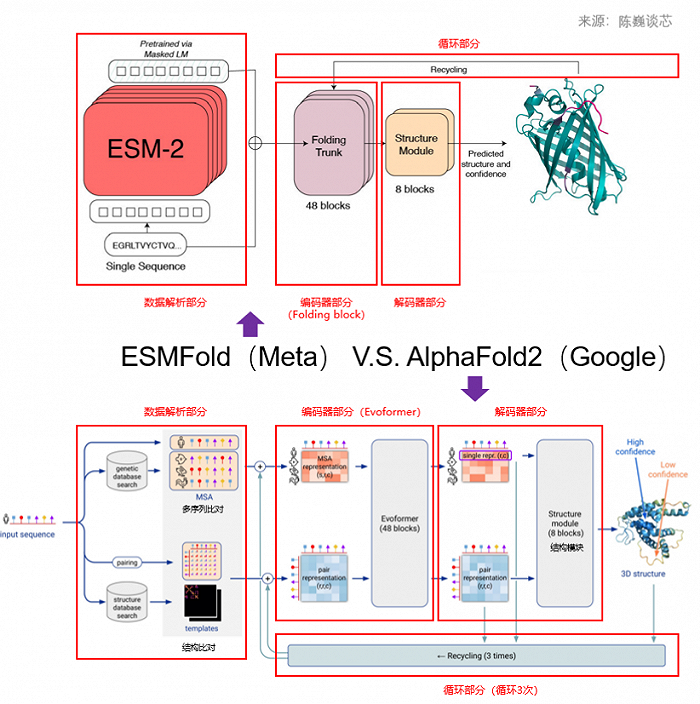
▲ESMFold与AlphaFold2对比
在AlphaFold2和RoseTTAFold中使用MSA和模板会导致两个瓶颈。
首先,可能需要基于CPU检索和对齐MSA和模板。这是由于AlphaFold2和RoseTTAFold不是二维序列嵌入状态,而是使用轴向注意力对应于MSA的三维内部状态进行操作,即使使用GPU,这一计算的代价也不菲。
相比之下,ESMFold是一个完全端到端的序列结构预测器,可以完全在GPU上运行,无需访问任何外部数据库。
例如在单个NVIDIA V100 GPU上,使用较少参数的ESMFold在14.2秒内对具有384个残基的蛋白质进行预测,可比单个AlphaFold2模型快6倍。而在较短的序列上,我们甚至看到了约60倍的改进。
速度的数量级提高是ESMFold优于AlphaFold2的独特优势,使我们能够在比现有方法更短的时间尺度内构建大量预测结构。考虑到可用序列数据的规模,这一点尤其重要。
例如,AlphaFold2蛋白质结构数据库的初始版本发布时具有约36万个预测结构,截至2022年7月则包含约99.5万个预测,这比目前许多蛋白质序列数据库小几个数量级。
04.数据解析部分与解码器的深度分析
数据解析部分用于输入序列和数据库的解析,为编码器提供输入。
在AlphaFold2模型中,数据解析部分使用了氨基酸序列数据库和结构数据库,分别用于相近序列的比对和结构模板的配对。
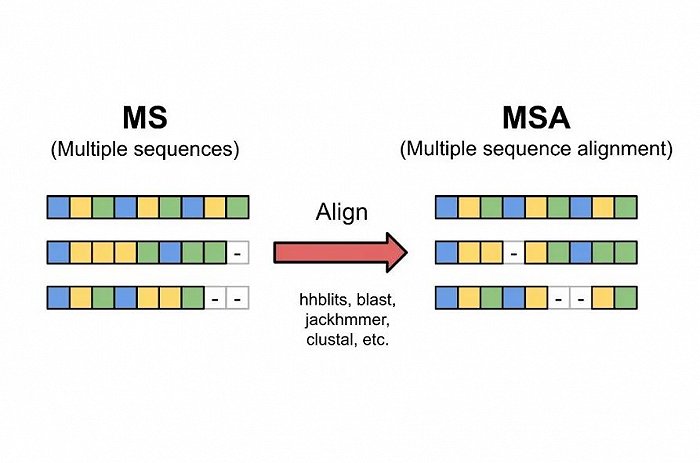
▲AlphaFold2多序列比对示意
生物信息学的基础是基于这样的一个假设:序列相似,结构相似,功能相似。一般认为相近的序列或者相近的结构会衍生出相近的功能域。
1)序列数据库被用于多序列比对(Multiple Sequence Alignment,MSA),即在序列数据库中检索与输入序列接近的数据库序列。
2)结构数据库则用于结构匹配,寻找与输入序列的结构接近的已知结构模板。
然后序列比对与结构比对的结果作为输入传输给编码器部分。
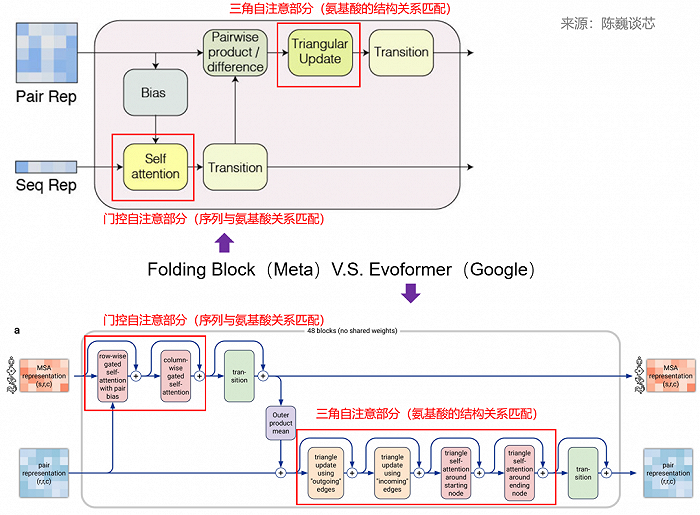
▲ESMFold Folding Block与AlphaFold2 Evoformer结构对比
解码器部分即Folding Trunk,一共48层。
ESMFold与AlphaFold2的一个关键区别是,ESMFold使用语言模型表示,消除了对明确的同源序列(以MSA的形式)作为输入的需要。
ESMFold通过用一个处理序列的Transformer模块取代处理MSA的计算昂贵的网络模块,简化了AlphaFold2中的Evoformer。这种简化意味着ESMFold的速度大大提高,远高于基于MSA的模型。
05.结语
作为蛋白质结构预测大模型,ESMFold获得准确原子分辨率结构预测的推断(Inferenc)速度比AlphaFold2提高了约一个数量级。特别是在实际计算中,这一速度优势表现的更加明显。这是由于ESMFold削减了搜索多序列来构建MSA的计算量。
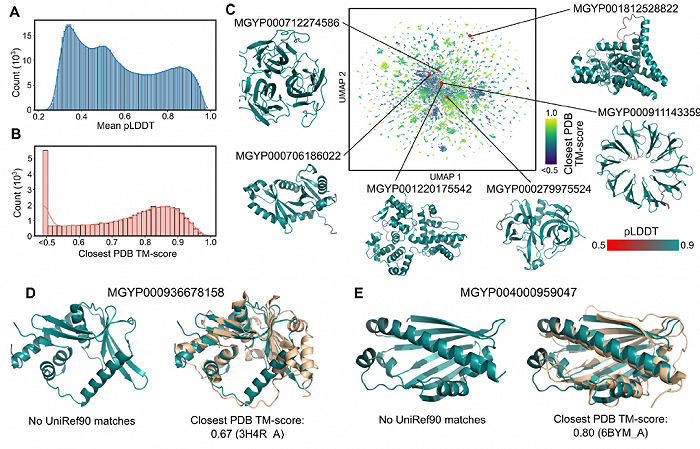
▲ESMFold用于探索宏基因组结构空间
推断速度优势使得基于计算有效映射大型宏基因组序列数据库的结构空间成为可能。
除了用于识别远同源性外,ESMFold还可以被用于进行快速准确的结构预测,并在实际时间尺度内获得数百万个预测结构,进一步帮助发现新的蛋白质结构和功能。这相当于在使用AI计算来构建生命的“元宇宙”。
150亿参数大模型,10x倍速度提升。虽然Meta ESMFold精度上没能做到全面“碾压”AlphaFold2,但毕竟“唯快不破”,对于蛋白质结构解析与预测、构建大型宏基因组结构数据库有着巨大的推动作用。
参考文献:
Zeming Lin et. al., Language models of protein sequences at the scale of evolution enable accurate structure prediction, https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
Jumper, J. et al., Highly accurate protein structure prediction with AlphaFold, Nature (2021):1-11.
评论