
文|氨基观察
天下武功,唯快不破。创新药研发领域,更是遵循此道。
不过,欲速则不达。在真正的临床试验中,往往会遇到这样或那样的问题,拖慢临床试验的进度。但这些问题,并不是都不可避免。
比如,礼来的阿尔茨海默症药物Donanemab,由于临床试验中“入组人数”少6名,加速审批遭到FDA拒绝。
这是一个本可以避免的临床错误,却给礼来造成数以亿计的损失,Donanemab也由此失去先发优势。
事实上,包括阿尔兹海默症在内的慢性病临床试验,由于临床规模大、跟踪随访时间长,对药企综合执行能力的要求越来越高。
不同的设计决定了一个临床试验的结果,而一个床试验的结果则决定了产品的走向。
要赢下这场竞赛,所有药企都要做得更多、更好。
01 阿尔茨海默症竞速赛,礼来落后
在研的阿尔茨海默症药物中,礼来的Donanemab一度备受期待。
原因在于,Donanemab与已获批上市的阿尔茨海默症药物都有得一拼。
与已获批上市的阿尔茨海默症药物Aduhelm相比,Donanemab在头对头临床试验中,显示出比Aduhelm更强的清除患者大脑中淀粉样蛋白的能力;而与另一款获批上市的阿尔茨海默症药物Lecanemab相比,Donanemab则与其作用机理类似。
在不少人看来,这两款药物都能获批上市,Donanemab获批上市的问题应该也不大。因此,市场对Donanemab期待颇高,Evaluate Pharma曾预测,Donanemab如果能够成功获批上市,2026年的销售额将达到31亿美元。
不过,市场对于Donanemab的美好畅想,夭折在了第一步。
1月19日,FDA拒绝了Donanemab用于治疗早期症状性阿尔茨海默病的加速批准。
理由很简单,不是因为安全问题,也不是因为效果问题,而是因为在临床试验中入组的人数不够。
在临床试验中,Donanemab试验组中的患者仅有94人,而FDA要求礼来提供至少100名接受过12个月持续治疗的患者数据。
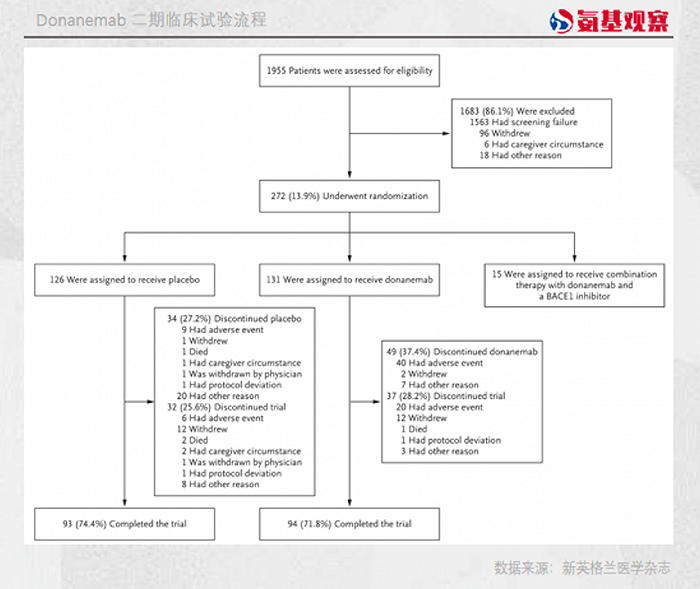
仅仅6名患者的缺失,给礼来造成的损失却要以亿为单位计算。这并非危言耸听,加速批准失败意味着Donanemab如果想要获批上市,需要等待完整的三期临床试验结果出炉。
礼来预计,符合FDA要求有100名患者参与的临床试验结果,将在2023年第二季度公布。届时,礼来需要凭借这份临床数据,再次向FDA递交药物上市申请,整个过程少说也要个一年半载。
耗钱耗时费力不说,更重要的是,Donanemab丢掉了先发优势。
原本Donanemab和Lecanemab应该可以前后脚获批上市,同台竞技。但如今,Donanemab上市还遥遥无期,Lecanemab则在1月6日成功获批上市,并开启商业化之路。
要知道,对于阿尔茨海默症这种慢性病来说,患者并不会轻易换药。这一年多的时间差距,已经足够Lecanemab提前占领医生和患者的心智。
在这场竞速赛中,礼来失去了先发优势。更令人惋惜的是,患者数量不足的问题,本可以在临床试验开展之前便解决的。
02 一个可以避免的临床错误
对于礼来来说,临床试验入组患者不足这个问题是完全可以避免的。
毕竟,在临床试验中入组多少患者这件事上,监管是有指导标准的。
目前,不管是FDA还是EMA,对入组患者数量及参与者的研究的时间长度要求,均遵循国际协调会议 (ICH) E1指南的建议:临床试验中患者总样本量至少为1000至1500人,用药6个月患者至少300人,用药12个月患者至少100人。
对于一些罕见病来说,药物临床门槛可能稍低,即便入组患者少于这一标准,也还有希望获批上市。
但对于阿尔茨海默症这种患者群体庞大的慢性病来说,临床试验中样本量要足够大才具备参考价值,才能进行长期跟踪研究。
一开始,Donanemab的入组患者数量也能满足FDA的要求。Donanemab的临床试验中,有257名患者入组,其中分到治疗组患者131名、安慰剂组有126名。
那么,试验做到最后,为什么又会出现患者不足的情况呢?
按照礼来的说法,患者不足是由于斑块减少导致许多患者在治疗6个月时就可以停止给药,最终导致只有不足100名患者接受Donanemab治疗时间达到了12个月。
当然,不管患者数量不足背后的原因究竟是什么,本质上,这都是一个本可以避免的事件。因为在临床试验中,药企都会提前预估患者退出率,在招募患者时按照推退出率提前算好盈余的患者样本量。
比如,Lecanemab在临床试验中,便预计18个月患者的退出率为20%,并提前做好了盈余的患者招募。
实际上,礼来阿尔茨海默症的竞品药物,在临床试验中的患者数量均不少。比如Lecanemab和Aduhelm。
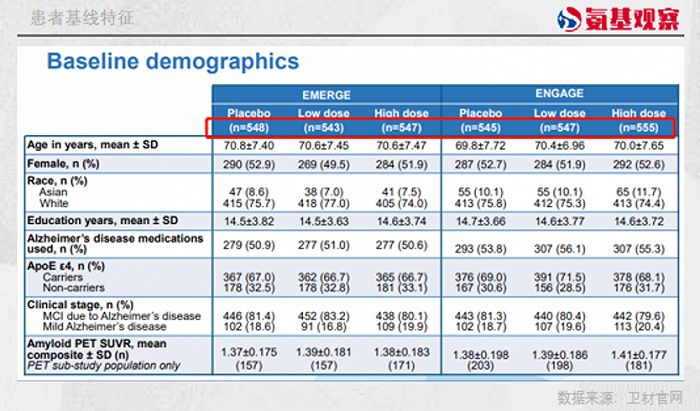
Aduhelm获批上市是基于一项名为EMERGE的三期临床试验。在这项临床试验中,安慰剂组、低剂量治疗组和高剂量治疗组患者数量都在500人以上。
Lecanemab的获批则是基于一项名为Clarity AD 的三期临床试验,这项临床实验共纳入了1795名患者。其中,安慰剂组和Lecanemab组患者各897人和898人,最终完成18个月治疗的患者,两组各有729人和757人。
相比之下,Donanemab最终仅94人完成12个月治疗的临床数据,也就显得不那么具备说服力。
这再次提醒我们,不同的设计决定了一个临床试验的结果,而一个临床试验的结果则决定了产品的走向。如果在当初临床试验设计中,礼来在招募患者时留出更多的盈余样本量,今日故事的走向或许会不一样。
03 来自慢性病临床试验的挑战
事实上,慢性病的特征,对药企的临床设计、执行力提出了更高的挑战。
正如前文所说,对于患者群体庞大的慢性病来说,临床试验中样本量要足够大才具备参考价值,也能尽可能避免患者脱落的影响。因此,慢性病临床规模动辄成千上万人。
比如阿尔茨海默病,目前全世界有超过5500万阿尔茨海默症患者。而前文提到的Aduhelm和Lecanemab,三期临床实验患者规模均在1500人以上。
再比如慢性体重管理。基于庞大的肥胖和更为庞大的爱美人群,减肥药(GLP-1受体激动剂)成了各大药企眼中的香饽饽。
不久前,诺和诺德开启一项GLP-1双靶点临床,入组患者达4000人;2018年,司美格鲁肽在超重/心血管适应症方面的临床,患者数量达17500人。礼来也不甘落后,去年10月开启了一项15000人的减肥大临床。
慢性病患者规模大的同时,异质性也较大。异质性指的是疾病的发病机制复杂,即便是相同的疾病的发病机制却可能并不相同。阿尔茨海默症最为典型,其诱发因素可以达到30多种,如年龄、遗传因素、脑部缺血缺氧、脑损伤等等。
面对这些异质性,只有当临床试验中纳入的样本量足够大时,才更可能保证各种类型的患者都会被纳入研究中,临床结果也就越可信。也就是统计学中的“大数原则”。
如果你想要做同一个适应症的临床试验,需要什么资源?
首先,很多钱和资源。
大规模临床试验,就是一个庞大的“吞金兽”。有钱还不是万能的。因为大型临床试验,不仅需要“买”大量的病人,更需要大量的优质的医生、研究员、牵头专家,而这些核心资源都是稀缺的。
其次,极强的随访能力。
慢性病临床试验,不仅规模大,还需要长时间跟踪患者,持续不断对这些患者进行随访、数据分析,如何减少患者在临床试验的脱落,如何保证临床试验的可持续性,同样是不小的挑战。
相比罕见病通常只有几百甚至几十人的患者入组规模,慢性病临床的难度无疑提高了N个量级。所以,大规模临床试验考验的是一家药企的综合执行能力。
当然,难度越大,也意味着一旦率先攻克,商业化“钱”景越大。
评论